Check out this interesting cartoon video on The Nephron
http://www.khanacademy.org/video/the-kidney-and-nephron

Sunday, August 29, 2010
Friday, August 27, 2010
CMV infection in Kidney Transplantation
Here is a brief presentation by Arun Chawla, MD on CMV in Kidney Transplantation.
CMV in Organ Transplantation
View more presentations from Nephrology, NSLIJ.
Thursday, August 26, 2010
IN THE NEWS: LIVER induced erythropoietin production
 As we all know that the kidney is a major site for erythropoietin production. We deal with complications of ESRD and patients needing EPO as a result.
As we all know that the kidney is a major site for erythropoietin production. We deal with complications of ESRD and patients needing EPO as a result. Apparently, liver controls production of this hormone in the fetal life. Hypoxa inducible factor of HIF inhibits this function of the liver by three hydroxylases PHD1,2 and 3.
This recent study listed below showed that in mice if all three PHD1,2,3 are inhibited, the liver can started producing erythropoietin again. This might make way for some clever drug creations for future treatment of anemia. The concern might be if the body chose to shut down liver mediated Epo, perhaps it might not be that active or perhaps not a safe form. No one knows.
Is liver produced epo as effective and safe as the kidney produced epo?
Regardless, worth a read!
Reference:
http://www.ncbi.nlm.nih.gov/pubmed/20651146
JOURNAL CLUB: RITUXIVAS Trial
We discussed the recent NEJM trial called RITUXIVAS
( Linked below)
Take home points
1. Randomized controlled double blinded trial that was comparing IV Cytoxan induction arm to IV Rituximab + low dose IV cytoxan arm and follow up for up to 12 months.
2. It was a random assignment via a 3:1 ratio ( rituximab to cytoxan)
3. everyone got steroids
4. Primary end points were sustained remission
5. Result: A rituximab-based regimen was not superior to standard intravenous cyclophosphamide for severe ANCA-associated vasculitis. Sustained-remission rates were high in both groups, and the rituximab-based regimen was not associated with reductions in early severe adverse events.
Plus points:
1.First study to compare SEVERE anca vasculitis in a randomized fashion that involves Rituximab
2. Patients included elderly as well which is where the choice of treatment becomes an issue.
Minus points:
1. Sample size
2. The Rituximab arm was sicker( more patients on dialysis)
3. The Rituximab arm received cytoxan as well ( unclear why they chose to do this, perhaps ethical reasons)
4. Net immunosuppresion was likely higher in the Rituximab arm ( dialysis , likely had TPE, and also cytoxan) making more infectious risks.
5. Data on IV cytoxan itself is poor in ANCA vasculitis.
6. Didn't really answer the question if Rituximab alone is good enough when compared to Cytoxan, and less toxic
http://www.ncbi.nlm.nih.gov/pubmed/20647198
( Linked below)
Take home points
1. Randomized controlled double blinded trial that was comparing IV Cytoxan induction arm to IV Rituximab + low dose IV cytoxan arm and follow up for up to 12 months.
2. It was a random assignment via a 3:1 ratio ( rituximab to cytoxan)
3. everyone got steroids
4. Primary end points were sustained remission
5. Result: A rituximab-based regimen was not superior to standard intravenous cyclophosphamide for severe ANCA-associated vasculitis. Sustained-remission rates were high in both groups, and the rituximab-based regimen was not associated with reductions in early severe adverse events.
Plus points:
1.First study to compare SEVERE anca vasculitis in a randomized fashion that involves Rituximab
2. Patients included elderly as well which is where the choice of treatment becomes an issue.
Minus points:
1. Sample size
2. The Rituximab arm was sicker( more patients on dialysis)
3. The Rituximab arm received cytoxan as well ( unclear why they chose to do this, perhaps ethical reasons)
4. Net immunosuppresion was likely higher in the Rituximab arm ( dialysis , likely had TPE, and also cytoxan) making more infectious risks.
5. Data on IV cytoxan itself is poor in ANCA vasculitis.
6. Didn't really answer the question if Rituximab alone is good enough when compared to Cytoxan, and less toxic
http://www.ncbi.nlm.nih.gov/pubmed/20647198
Interferon Gamma use in fungal infections in transplant patients
A recent article in AJT reports the use of Interferon Gamma as a salvage and additional therapy for disseminated fungal infections in kidney transplant recipients.
We always get concerned with giving interferons to transplant recipients as it will increase and promote an immune response and pose a risk of rejection.
In the above cases, not only did the patients survived but the graft was not effect in 4/7 cases as well with no consequences of rejection. Why interferon gamma? It is the most important cytokine in communication of T cells to fight fungal and mycobacterial infections.
The idea is novel yet scary. More data and more cases are needed till we think of possibly using this in large clinical settings. Clearly, the concern of activating more crosstalk among T helper cells by using Interferon Gamma is helpful in fighting infection but might have to be the right balance to prevent rejection.
Reference:
http://www.ncbi.nlm.nih.gov/pubmed/20353472
http://www.ncbi.nlm.nih.gov/pubmed/20636453
We always get concerned with giving interferons to transplant recipients as it will increase and promote an immune response and pose a risk of rejection.
In the above cases, not only did the patients survived but the graft was not effect in 4/7 cases as well with no consequences of rejection. Why interferon gamma? It is the most important cytokine in communication of T cells to fight fungal and mycobacterial infections.
The idea is novel yet scary. More data and more cases are needed till we think of possibly using this in large clinical settings. Clearly, the concern of activating more crosstalk among T helper cells by using Interferon Gamma is helpful in fighting infection but might have to be the right balance to prevent rejection.
Reference:
http://www.ncbi.nlm.nih.gov/pubmed/20353472
http://www.ncbi.nlm.nih.gov/pubmed/20636453
Wednesday, August 25, 2010
IN THE NEWS- Hyperkalemia review
 The most recent AJKD 2010 issue has a more physiologic approach to diagnosis of hyperkalemia. The picture on the left from the AJKD article is a must look at! It says a thousand words. When we think of causes of hyperkalemia( not pseudohyperkalemia), if we start from the top with RENIN production to its effect on ANG 1 and then ANG II and Aldosterone to the effects on the principal cell, you won't miss a cause.
The most recent AJKD 2010 issue has a more physiologic approach to diagnosis of hyperkalemia. The picture on the left from the AJKD article is a must look at! It says a thousand words. When we think of causes of hyperkalemia( not pseudohyperkalemia), if we start from the top with RENIN production to its effect on ANG 1 and then ANG II and Aldosterone to the effects on the principal cell, you won't miss a cause.Either one of these pathways can be altered leading to hyperkalemia!
References:
http://www.ncbi.nlm.nih.gov/pubmed/20493606
http://www.ncbi.nlm.nih.gov/pubmed/20570423
Quiz 4 Answers
What is the most common cause of Denovo Thrombotic Microangiopathy post renal transplant?
Calcineurin inhibitor toxicity 5 (55%)
Antibody mediated rejection 3 (33%)
MPGN 0 (0%)
Malignancy 0 (0%)
Infections 1 (11%)
Denovo TMA post transplant is defined as happening in the early post transplant period.( 6 months). All of the above can cause de novo TMA in post transplant patients. The most common cause as most of you got it is CNI toxicity. Although, Antibody mediated rejection closely follows it and should always be considered. Another cause not listed here is ischemic repefusion injury as well leading to a TMA.
A recent study in AJT August 2010 showed in a retrospective fashion that TMA can be very strongly associated with C4D positive Biopsies showing ABMR as well. 14% with CD4 positive patients had TMA compared to only 3% with C4D negative biopsies. Treatment changes - Plasmapheresis might help in this case as you will remove the donor specific antibodies and also help the process of TMA treatment.
Regardless in any case of TMA, treat the underlying cause if found! CNI induced TMA doesn't NEED to be treated with plasmapheresis
References:
http://www.ncbi.nlm.nih.gov/pubmed/20659088
Calcineurin inhibitor toxicity 5 (55%)
Antibody mediated rejection 3 (33%)
MPGN 0 (0%)
Malignancy 0 (0%)
Infections 1 (11%)
Denovo TMA post transplant is defined as happening in the early post transplant period.( 6 months). All of the above can cause de novo TMA in post transplant patients. The most common cause as most of you got it is CNI toxicity. Although, Antibody mediated rejection closely follows it and should always be considered. Another cause not listed here is ischemic repefusion injury as well leading to a TMA.
A recent study in AJT August 2010 showed in a retrospective fashion that TMA can be very strongly associated with C4D positive Biopsies showing ABMR as well. 14% with CD4 positive patients had TMA compared to only 3% with C4D negative biopsies. Treatment changes - Plasmapheresis might help in this case as you will remove the donor specific antibodies and also help the process of TMA treatment.
Regardless in any case of TMA, treat the underlying cause if found! CNI induced TMA doesn't NEED to be treated with plasmapheresis
References:
http://www.ncbi.nlm.nih.gov/pubmed/20659088
The Kidney is not silent
"Many doctors may view nephrology as a remarkable kind of intensive care. A patient presents in an acute crisis, close to death. The creatinine is over a thousand. Immediate transfer to the renal team follows, then the magical effects of dialysis, and finally recovery. Nephrology is not for normal doctors. The kidney is for exceptionally skilled specialist physicians." -- LANCET
Check out this exclusive editorial regarding Renal as a field in medicine
http://www.thelancet.com/journals/lancet/article/PIIS0140673610605247/fulltext?rss=yes
Check out this exclusive editorial regarding Renal as a field in medicine
http://www.thelancet.com/journals/lancet/article/PIIS0140673610605247/fulltext?rss=yes
Saturday, August 21, 2010
CLINICAL CASE 24, ANSWERS and SUMMARY
Name of the odd one out
Dense Deposit Disease 0%
Immunotactoid GN 66%
Familial MPGN 3 11%
C5HR5 Nephropathy 16%
C3 Glomerulonephritis 5%
Most of you got this one right. This is in light with the new and upcoming potential new classification of GNs under a C3 Glomerulopathies. All except Immunotactoid GN is considered a C3 glomerulopathy. Immunotactoid GN would fall under a class of GN with organized deposits and under types such as fibrillary, amyloid and paraproteinemias.
I think its about time that most of us start thinking of MPGN as a pattern of injury rather than a disease till there are deposits. What the C3 glomerulopathy classification is trying to tell us is that the injury is due to the problem in the complement cascade and hence leading to different disease forms that ultimately might lead to double contouring and MPGN like lesions.
What constitutes C3 glomerulopathies: Glomerular deposits of complement C3 and absence of immunoglobulin within glomeruli or just pauci immunoglobin deposition.
Examples of these diseases are: Dense deposit disease, Idiopathic C3 glomerulopathy, MPGN type 1 with just isolated complement C3 deposits, Familial MPGN III, CFHR5 Nephropathy.
1. Idiopathic MPGN1: Usually light showing MPGN patten and immuno with IgG and C3 staining and no secondary cause identified
2. DDD: Dense deposits that are intramembranous and diffuse C3 staining and no immunoglobulin staining. associated with C3 nephritic factor problem
3. C3 Glomerulonephritis: subendothelial and mesangial deposits of just isolated C3 staining. Possible complement dysregulation
4. Familial MPGN3: subepithelial and subendothelial deposits , linked to chromosome 1 leading to Factor H related problem
5. CFHR5 Nephropathy: Like C3 GN but have a Factor H related mutation encoded by the CFHR5 gene.
Why are these classification important: perhaps we can look for these causes in those specific cases and perhaps even help in potential donor choices for transplantation. Medications affecting the complement system such as eczulimab might be of benefit in some of these disease entities in the near future.
If one suspects C3 glomerulopathies: its worth checking the complement cascade function: C3, C4, Factor H and I, B levels. Check also for Nephritic factor C3, Factor H antibodies, CD46 quantification.
Certain genetic mutations screenings are also available.
Below is a nice review in Nature Review Nephrology. There are nice pathology pictures and review tables.
Have a nice read.
http://www.ncbi.nlm.nih.gov/pubmed/20606628
http://www.ncbi.nlm.nih.gov/pubmed/17018561
http://www.ncbi.nlm.nih.gov/pubmed/19190809
Dense Deposit Disease 0%
Immunotactoid GN 66%
Familial MPGN 3 11%
C5HR5 Nephropathy 16%
C3 Glomerulonephritis 5%
Most of you got this one right. This is in light with the new and upcoming potential new classification of GNs under a C3 Glomerulopathies. All except Immunotactoid GN is considered a C3 glomerulopathy. Immunotactoid GN would fall under a class of GN with organized deposits and under types such as fibrillary, amyloid and paraproteinemias.
I think its about time that most of us start thinking of MPGN as a pattern of injury rather than a disease till there are deposits. What the C3 glomerulopathy classification is trying to tell us is that the injury is due to the problem in the complement cascade and hence leading to different disease forms that ultimately might lead to double contouring and MPGN like lesions.
What constitutes C3 glomerulopathies: Glomerular deposits of complement C3 and absence of immunoglobulin within glomeruli or just pauci immunoglobin deposition.
Examples of these diseases are: Dense deposit disease, Idiopathic C3 glomerulopathy, MPGN type 1 with just isolated complement C3 deposits, Familial MPGN III, CFHR5 Nephropathy.
1. Idiopathic MPGN1: Usually light showing MPGN patten and immuno with IgG and C3 staining and no secondary cause identified
2. DDD: Dense deposits that are intramembranous and diffuse C3 staining and no immunoglobulin staining. associated with C3 nephritic factor problem
3. C3 Glomerulonephritis: subendothelial and mesangial deposits of just isolated C3 staining. Possible complement dysregulation
4. Familial MPGN3: subepithelial and subendothelial deposits , linked to chromosome 1 leading to Factor H related problem
5. CFHR5 Nephropathy: Like C3 GN but have a Factor H related mutation encoded by the CFHR5 gene.
Why are these classification important: perhaps we can look for these causes in those specific cases and perhaps even help in potential donor choices for transplantation. Medications affecting the complement system such as eczulimab might be of benefit in some of these disease entities in the near future.
If one suspects C3 glomerulopathies: its worth checking the complement cascade function: C3, C4, Factor H and I, B levels. Check also for Nephritic factor C3, Factor H antibodies, CD46 quantification.
Certain genetic mutations screenings are also available.
Below is a nice review in Nature Review Nephrology. There are nice pathology pictures and review tables.
Have a nice read.
http://www.ncbi.nlm.nih.gov/pubmed/20606628
http://www.ncbi.nlm.nih.gov/pubmed/17018561
http://www.ncbi.nlm.nih.gov/pubmed/19190809
Friday, August 20, 2010
B cell and long term graft function
A lot of transplant patients do so well and require very minimal amounts of medications. What is the magic recipe for that? Perhaps it is T regs cells and amount of T regs vs T effector cells. Perhaps it is a subset of B cells.
A recent paper in Kidney International addresses this topic.
The investigators compared B cells of people with stable graft function and ones with chronic rejection and healthy volunteers. They found that the ones who had stable graft function with minimal drugs had increased B cells of activated, memory and early memory type. They had a enriched transcriptional profiling. The costimulatory molecules like CD40 and CD80 ligand were upregulated in these B cells. These cells were also giving out an inhibitory signal for proliferation and a preventive signal for Hyperactive B cell response. They expressed CD1D and CD5 ( another recent paper had suggested this as well).
This suggests that there a specific type of B cells out there that patients with good graft function and minimal drugs have that might be regulatory in nature and allow long term graft functioning.
More studies should be done to look out for these type of cells
http://www.ncbi.nlm.nih.gov/pubmed/20531452
Also, perhaps using anti co stiumatory blockades might not be all that ideal then? Perhaps. not much data to say yes or no!
A recent paper in Kidney International addresses this topic.
The investigators compared B cells of people with stable graft function and ones with chronic rejection and healthy volunteers. They found that the ones who had stable graft function with minimal drugs had increased B cells of activated, memory and early memory type. They had a enriched transcriptional profiling. The costimulatory molecules like CD40 and CD80 ligand were upregulated in these B cells. These cells were also giving out an inhibitory signal for proliferation and a preventive signal for Hyperactive B cell response. They expressed CD1D and CD5 ( another recent paper had suggested this as well).
This suggests that there a specific type of B cells out there that patients with good graft function and minimal drugs have that might be regulatory in nature and allow long term graft functioning.
More studies should be done to look out for these type of cells
http://www.ncbi.nlm.nih.gov/pubmed/20531452
Also, perhaps using anti co stiumatory blockades might not be all that ideal then? Perhaps. not much data to say yes or no!
Renal Fellow Network: Hot peppers for hypertension?
Renal Fellow Network: Hot peppers for hypertension?: "An interesting article was published in the August 4th edition of Cell Metabolism. I'm always intrigued when common food ingredients are us..."
Wednesday, August 18, 2010
CONSULT ROUNDS: LOW POTASSIUM STORY!
 When you encounter a hypokalemia with metabolic alkalsosis that is normo-tensive, there are very few diagnosis to consider: Diuretic use, abuse or diuretic like syndromes ( namely Bartter Syndrome(BS) and Gitelman's Syndrome(GS))
When you encounter a hypokalemia with metabolic alkalsosis that is normo-tensive, there are very few diagnosis to consider: Diuretic use, abuse or diuretic like syndromes ( namely Bartter Syndrome(BS) and Gitelman's Syndrome(GS))Take home points:
1. There are 4 different types of Bartter syndrome( basically anyway the cell in the thick ascending loop of henle can be affected in absorption of Na-Cl.)- a loop diuretic like effect
Bartter syndrome I - Defects in the Na-K-2Cl transporter
Bartter syndrome II - Defects in the apical potassium channel, caused by mutations in the ROMK1 gen
Bartter syndrome III - This is due to mutations in the CLCNKB gene leading to Cl channel problem in the basolateral surface.
Bartter syndrome IV - This is due to mutations in the CLCNKA gene. Again affecting the Cl-K exchange in the basolateral surface.
2. A fifth variant might occur that is AD as opposed to being AR (the above 4 variants) that affects the Ca sensing receptor in the basolateral surface.
3. Most of the above lead to hypocalcemia and hypercalciuria.
4. Gitelman's syndrome is a loss-of-function mutations in the SLC12A3 gene that codes for the thiazide-sensitive Na-Cl cotransporter in the distal convoluted tubule. This leads to a more pronounced hypomagnesemia and hypocalciuria compared to the prior. This is a more common entity in adults
5. An interesting way to differentiate the two would be to use diuretics to observe the Urinary Cl and Na concentrations. A patient with GS, when given Thiazides will have no major change as that channel is already not working but will have an increase in Urinary CL and Na when given a Loop diuretic. Vice versa, the effect of loop of a patient with BS will show no changes but when given a thiazide will have increase urinary CL and Na.
6. Treatment is usually using K sparing agents like Amiloride, aldosterone antagonists or NSAIDs.
7. Chronic Hypokalemia can lead to hypokalemic related Nephropathy and ESRD.
8. Renin and Aldosterone levels are usually high in these cases.
Few good references regarding the use of diuretics to make diagnosis
http://www.ncbi.nlm.nih.gov/pubmed/17699451
http://www.ncbi.nlm.nih.gov/pubmed/1731022
http://www.ncbi.nlm.nih.gov/pubmed/12172059
Tuesday, August 17, 2010
IN THE NEWS: MIDODRINE
Midodrine is an oral alpha agonist that helps prevent intra dialytic hypotension.
What is the evidence that midodrine works for hypotension? Is there any? A nice review was published in NDT few years ago. Here is the summary. A meta analysis was done.
Observational studies, randomized controlled trials, crossover studies and pre- and post-intervention design studies with dialysis patients were included. Thirty-seven full text articles were retrieved and nine met the selection criteria, in addition to one unpublished study. Midodrine dosing regimens ranged from 2.5 to 10 mg of midodrine given 15–30 min pre dialysis. Post-dialysis systolic blood pressure was higher by 12.4 mmHg and diastolic pressure was higher by 7.3 mmHg during treatment vs. control. Six of 10 studies report improvement in symptoms of IDH, and there were no reported serious adverse events ascribed to midodrine.
This review points out that midodrine has a role in the therapy of hemodialysis patients experiencing IDH. Although not any of these studies had that big of a sample size. FDA just issued a warning that this drug be take away from the market. This is due to lack of Class I or II evidence for overall treatment of cardiac and renal diseases. Check out the FDA website.
Here is the review to the article
http://www.ncbi.nlm.nih.gov/pubmed/15280522
http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm222580.htm?utm_source=twitterfeed&utm_medium=twitter
Monday, August 16, 2010
CONSULT ROUNDS: ANCA negative Pauci-Immune Cresentric Glomerulonephritis
ANCA negative PICGN is present in 10% - 30% of all Type III RPGN (type I being Anti GBM and type II being Immune complex associated like lupus).
They typically have fewer extrarenal symptoms than patients with ANCA positive GN.
Patients with ANCA negative disease have been described to have higher proteinuria than their counterparts.
Pathogenesis is unclear but neutrophils leading to respiratory burst and free radical injury are quintessential; how they are stimulated is a matter of debate. Anti endothelial cell antibodies (AECA) and Anti lysosomal protein antibody are some of the other antibodies that can stimulate neutrophils but they are also present in ANCA pos patients. Role of cell mediated immunity is also being studied.
Treatment modalities are similar and outcomes are comparable. Some of the major studies looking at ANCA negative PICGN are summarised below -
http://www.ncbi.nlm.nih.gov/pubmed/19662604
Transplantatation of two kidneys in marginal donors
http://nephrohug.com/2010/08/16/greffe-des-deux-reins/
Check out the above French Blog on this topic from Nephrohug.
Check out the above French Blog on this topic from Nephrohug.
Friday, August 13, 2010
The Micro RNA blog
Take a look at this extensive summaries of blogs just on micro RNAs
Very neat blog
http://mirnablog.com/
Very neat blog
http://mirnablog.com/
Thursday, August 12, 2010
.JPG)
CONSULT ROUNDS: Resp Alkalosis
Here is an ABG: 7.71/17/118 and on Room Air and Hco3 is 22.
At a glance this is respiratory and metabolic alkalosis. This can be possible especially in someone on Peritoneal Dialysis. The compensation of Resp Alkalosis is dumping bicarbonate in the urine; but if you don't make urine and are on PD- more alkaline fluid:- this is very possible.
Lets review the causes of Resp Alkalosis: Four major causes that can include the rest
Stimulation of the resp center, Hypoxemia, Pulmonary or cardiac disease and finally ventilators.
So a complete list would be CNS disorders, endotoxins( sepsis), Anxiety, fever, ASA toxicity, anything that increases A-a gradient( pulm embolism, edema, Pneumonia), cardiac disease( MI), pregnancy, liver disease, progesterone, and some toxins ( medications).
A ph >7.6 puts an increase risk of seizures. Treat the underlying cause but try to suppress the resp drive with anxiolytics, sedatives or bagging or if need be Intubation.
What other situations you get a met alk and resp alk together?
Liver disease with diuretics; Pregnancy with vomiting are other examples
Wednesday, August 11, 2010
BKV viral protein-1 mRNA in urinary cells
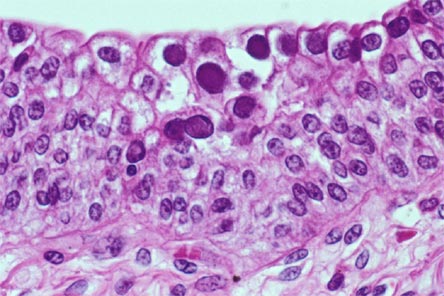
A noninvasive, accurate biomarker for diagnosis of BKVN is being sought. Recent article in Transplantation reports using the urinary cell mRNA profile at the time of BK Nephropathy diagnosis and compared to risk of graft function.
BK Nephropathy was diagnosed with a sensitivity of 100% and specificity of 97% using the urinary mRNA. Levels of granzyme B (GB) mRNA and proteinase inhibitor (PI)-9 mRNA in urinary cells were higher in BKV patients with a subsequent decline in renal function compared with patients with stable function, and were positively associated with rise in serum creatinine from the time of BK diagnosis to 12 months after diagnosis.
This confirms the fact that urinary granzyme B and PI-9 could be used as markers of inflammation during any renal episode post transplant. Similar findings were seen in rejection as well. It seems that no matter what causes the inflammation, BK or rejection, a rise in urinary GB and PI-9 suggests a poor prognosis.
Reference:
TOPIC DISCUSSION: Plasma Pheresis and Renal Disease

Plasmapheresis appears to be a useful adjunct to conventional therapy in the treatment of anti-GBM nephritis, severe dialysis-dependent forms of pauciimmune RPGN, cryoglobulinemia, and HUS-TTP, some severe cases of cast nephropathy and in antibody mediated rejection, desensitization and ABOI transplants. But data from controlled trials do not support a role for plasmapheresis in immune complex-mediated RPGN, such as lupus nephritis, and acute allograft rejection. Although Lupus Nephritis with TTP or TMA might benefit from TPE.
While data on use of Plasma Pheresis or Apheresis ( TPE) is robust in neurological and hematological diseases, the clinical efficacy of plasmapheresis for many acute renal conditions is still controversial.
While data on use of Plasma Pheresis or Apheresis ( TPE) is robust in neurological and hematological diseases, the clinical efficacy of plasmapheresis for many acute renal conditions is still controversial.
The three classic diseases that is worth discussing are ANCA RPGN, anti GBM and
Cast Nephropathy. Lupus Nephritis is also mentioned.
The data on ANCA RPGN is as follows: Based on a recent trial in Europe, the role of TPE in ANCA RPGN is when the crt is >5.7 or they are dialysis dependent. It appears that TPE offers some benefit of disease reversal and better renal outcomes if employed in dialysis dependent forms of the disease.
The data on ANCA RPGN is as follows: Based on a recent trial in Europe, the role of TPE in ANCA RPGN is when the crt is >5.7 or they are dialysis dependent. It appears that TPE offers some benefit of disease reversal and better renal outcomes if employed in dialysis dependent forms of the disease.
On the other hand, Anti GBM disease offers the MOST benefit when employed at a time when the patient is NOT dialysis dependent. If they are dialysis dependent, the prognosis of disease doesn't change much with TPE. Pulmonary hemorrhage would be other indication.
In terms of Cast Nephropathy, it is controversial. A study in 2005 showed that patients with cast nephropathy benefited from few sessions of TPE. This study was scrutinized as many of the patients were never biopsied, so we don't know for sure we are treating cast nephropathy. At this point, perhaps a severe cast nephropathy biopsy proven on dialysis might benefit from TPE but there is no concrete evidence.
What about lupus Nephritis? The investigators in 2005 found, based upon a review of clinical trials, that the current use of cyclophosphamide combined with steroids remains the best option to preserve renal function in proliferative lupus nephritis. The authors found no benefit with addition of plasma exchange to cyclophosphamide or azathioprine plus steroids for mortality or end-stage renal failure. Some anecdotal evidence suggests the use of TPE in Lupus with TMA.
References:
Monday, August 9, 2010
IN THE NEWS: DETECTIVE NEPHRON's NEXT VENTURE

http://www.asn-online.org/publications/kidneynews/archives/2010/aug/KN_aug2010.pdf
This month's ASN Kidney News 2010 August has the next adventure of the renal detective.
He takes on a case of metabolic alkalosis. Check it out, pages 16-18.
CLINICAL CASE 23 , ANSWER and SUMMARY
A 56 y old male comes in with SIADH and a Na of 100, active seizures. You plan to give 3%. How would you give it?
Give 100ml of 3%Nacl bolus and repeat it maximum of 3 times within the first hour 38%
Give 3%Nacl at a faster rate of about 100cc/hr till Na is around 114 and then correct 0.5Meq/L for the remainder of the 24 hours 29%
Give 3%Nacl at around 60cc.hr and correct 0.5meq/L for the 24 hours 28%
Give v2 receptor antagonist 3%
Give 2%Nacl at 200cc/hr for 24 hours 0%
Interesting break down as expected. Majority of you are split between given 3% as bolus vs. initial accelerating and then slowing down vs. just picking one rate and going with it. The change in the paradigm of treating ACUTE symptomatic hyponatremia has been in the last few years. Hypertonic saline is warranted in someone who is seizing and has a Na that is low; since if not corrected, brain herniation can occur.
Choice no 1 was initially described in hyponatremic athletes participating in endurance events such as marathon races. It consists of 100 mL of 3 percent saline given as an intravenous bolus, which should acutely raise the serum sodium concentration by 2 to 3 meq/L, thereby reducing the degree of cerebral edema; if neurologic symptoms persist or worsen, a 100 mL bolus of 3 percent saline can be repeated one or two more times at 10 minute intervals The rationale for this approach is that, in patients with symptomatic hyponatremia, rapid increases in serum sodium of approximately 4 to 6 meq/L can reverse severe symptoms such as seizures. If you use this approach, you might not need to do much for the remaining time over 24 hours and patient can start self correcting.
Giving a bolus of 3% is not agreed upon by many Nephrologist. Take a look at the recent review that I pubmed below for a nice algorithm. The usual goals for the overall rate of correction are to raise the serum sodium less than 10 meq/L in the first 24 hours and less than 18 meq/L in the first 48 hours. Choice B which is giving a drip of 3%Nacl initially at a fast rate should also work in such instances and perhaps physicians might feel more comfortable using that approach. The rate of 3% given in the third choice might be too slow for someone who is symptomatic.
So the most accurate answer is Choice A, but as you can see, there is no hard data and people might use diverse options( B>C as the next best answer). Giving a bolus of 3%NACL – where is this data coming from? When looked at Neurosurgical patients with cerebral edema who were normonatremic, a 30cc bolus of 23.4% saline increased the Na by 5mmol/L and there was an excellent and quick reversal of herniation. A 30cc of 23.4% saline is equal to 240cc of 3% saline. Based on these findings and a recent study on exercise induced hyponatremia in marathon runners, the International Exercise Associated Hyponatremia consensus development conference recommends that any athlete with hyponatremia and acute changes that are symptomatic ( seizures, encephalopathy) should be treated with 100ml of 3% NACL to acutely reduce brain edema. Two additional doses can be given over a 10 minute interval if there is no improvement. Experts think that this approach is reasonable in any case of SEVERE ACUTE SYMPTOMATIC hyponatremia. Get the symptoms better, and then when you get to safe zone, you can correct more slowly or apply brakes with D5W or ddavp if need be. If the cause was acute, rapid correction is warranted. Chronic hyponatremia is a different story and asymptomatic acute is also a different story.
Take a look at these excellent references.
TOPIC DISCUSSION: Hyperkalemia due to cell shifts?
It is always important to make sure that K disorders such as hyper and hypokalemia are not due to cell shifts and Pseudo in nature. Cell shifts usually lead to more of hyperkalemia than hypokalemia. 2% shift in intracellular K to the ECF will result in serum K level of 8meq/L.
What causes Hyperkalemia due to cell shifting
Here is a comprehensive list that is worth looking for ( check out the recent AJKD listed below)
1. Insulin deficiency
2. Acidosis
3. Beta Blockade
4. Hypertonicity
5. Alpha stimulation
6. Rhabdomyolysis
7. Hemolysis
8. Tumor Lysis
9. Periodic Paralysis
10. Digoxin
11. Succinylcholine
12. Rebound after insulin infusion
http://www.ncbi.nlm.nih.gov/pubmed/20493606
What causes Hyperkalemia due to cell shifting
Here is a comprehensive list that is worth looking for ( check out the recent AJKD listed below)
1. Insulin deficiency
2. Acidosis
3. Beta Blockade
4. Hypertonicity
5. Alpha stimulation
6. Rhabdomyolysis
7. Hemolysis
8. Tumor Lysis
9. Periodic Paralysis
10. Digoxin
11. Succinylcholine
12. Rebound after insulin infusion
http://www.ncbi.nlm.nih.gov/pubmed/20493606
Sunday, August 8, 2010
Thursday, August 5, 2010
CLINICAL CASE 22, ANSWERS AND SUMMARY
A 60 y old male presents for the third time with edema, hypoalbuminemia, lower extremity edema and hypotension. Per patient, the episodes are cyclical and come and go. You diagnose Systemic Capillary Leak Syndrome? In which phase do you see renal failure?
Shock Phase 44%
Leak Phase 32%
Post Leak Phase 24%
Systemic capillary leak syndrome is a extremely rare disease that leads to vascular collapse. A classic place where one can remember seeing this is post IL-2 Infusion for renal cell cancer patients. This vascular collapse leads to hemoconcentration and low albumin levels. So far around 150 cases have been reported. In this entity, its important to differentiate this disease from Nephrotic Syndrome, Protein losing enteropathy, Carcinoid, Mastocytosis, Pheochromocytoma, Sepsis and Toxic Shock syndrome in the right clinical context.
In terms of pathophysiology, there is some data suggesting that the damage is due to endothelial dysfunction. But there are association studies also suggesting that 80% or so of patients with this syndrome get MGUS or have MGUS as well and there has been association with plasma cell dyscrasias.
In terms of renal disease and acute kidney injury in these patients: although there can be renal insult possible at any phase during this disease, the most likely phase is Shock Phase, as most of you suggested in your answer.
Typically, in this syndrome, there is a prodrome of weakness that leads to severe pre syncope, hemoconcentration, cool skin and oliguria early on and then there is the SHOCK PHASE that is usually causing pre renal/ATN leading to renal injury, stroke or DVTs. Within hours, this can progress to LEAK PHASE where edema develops along with low albumin and all extremities and face swell up. This leads to compartment syndrome and Rhabdomyolysis as well( could lead to renal injury here as well). Within 3 days, you get the POST LEAK PHASE, where there is restoration of volume and diuresis-- sometimes leading to cardiopulmonary failure ( could have renal injury here from post diuresis as well and cardiac failure)
A rare disease but usually we see renal failure with it. Typically in the SHOCK phase but other phases possible depending on the severity of it
A nice review in Annals of Internal Medicine
Check it out
Medical Innovation
Check out this nice website on medical education by NSLIJ Medicine
http://www.medicaleducationfoundation.com/
http://www.medicaleducationfoundation.com/
Wednesday, August 4, 2010
CONSULT ROUNDS: Management of BEER POTAMANIA

A nice algorithm is listed below; from an AJKD review recently on this topic:
Usually NPO in the first 24 hours
No IVFs unless symptomatic( VERY IMPORTANT), as they might correct very fast with just Normal Saline as you are increasing their solute load.
ICU admission
Keep checking Na every 2-3 hours
Correct rapid to 1-2meq in one hour if symptomatic else <<< 10 Meq/L in first 24 hours( may not need to do anything, usually self correct)
Relower serum sodium levels if necessary
Give all IV medications and nutrition via D5W only given their rapid correction rate
Low threshold to image the Brain.
Source: AJKD Beer potamania article
Reference:
http://www.ncbi.nlm.nih.gov/pubmed/17900468
Low Donor Kidney Weight ? Does it matter?
We have asked this question many times in conferences and especially when a pediatric kidney gets put in an adult recipient? Does it matter if the donor kidney weight is not compatible to the recipient's weight?
There is one study in 2005 that showed that low donor kidney weight to recipient weight ratio did not affect graft survival. That study only had 2.5 year follow up.
A recent study in JASN looked at >1000 patients for over 10 years and max of 7 year follow up. They found that with a low donor kidney to recipient kidney ratio of <2.3g/kg, initially the GFR increased, plateaued at 6 months but then decreased rapidly after 7 years at a mean rate of 3 ml/min.
With patients with >2.3g/kg, the the GFR after 7 years decreased at a much slower rate- 1.34ml/min.
The proteinuria was also higher in the low ratio group.FSGS was more common in the biopsies of the low ratio group.This is a retrospective analysis with its usual flaws but raises an important point of avoiding kidney and recipient weight incompatibility to avoid late clinical outcomes.
References:
http://www.ncbi.nlm.nih.gov/pubmed/20488949
http://www.ncbi.nlm.nih.gov/pubmed/15563571
There is one study in 2005 that showed that low donor kidney weight to recipient weight ratio did not affect graft survival. That study only had 2.5 year follow up.
A recent study in JASN looked at >1000 patients for over 10 years and max of 7 year follow up. They found that with a low donor kidney to recipient kidney ratio of <2.3g/kg, initially the GFR increased, plateaued at 6 months but then decreased rapidly after 7 years at a mean rate of 3 ml/min.
With patients with >2.3g/kg, the the GFR after 7 years decreased at a much slower rate- 1.34ml/min.
The proteinuria was also higher in the low ratio group.FSGS was more common in the biopsies of the low ratio group.This is a retrospective analysis with its usual flaws but raises an important point of avoiding kidney and recipient weight incompatibility to avoid late clinical outcomes.
References:
http://www.ncbi.nlm.nih.gov/pubmed/20488949
http://www.ncbi.nlm.nih.gov/pubmed/15563571
Renal Biopsy simulation
https://www.pediatric-nephrology.com/daily-updates/2010/08/04/259-kidneybx.html
Check out the above blog on Renal Biopsy simulation!
Check out the above blog on Renal Biopsy simulation!
Tuesday, August 3, 2010
Post transplant TMA, revisiting Atypical HUS

Post Transplantation is a real entity. Many causes have been identified. CNI toxicity, Sirolimus, ischemia, antibody mediated rejection or de novo carcinoma, antiphospholipid syndrome, post transplant SLE are a few possible diagnosis. One study showed that among 24 patients with post transplant TMA that was de novo, 7 carried a mutation in CFH or CFI or combined mutation, indicating a genetic abnormality that might be the first hit.
In the past decade, work has been very active in the field of Atypical HUS. Many complement abnormalities have been identified namely the CFH , CFI mutations, C3 mutations, CFB mutations, all which are mutations in the alternative pathway of complement leading to activation of MAC and TMA
A recent review in AJT July 2010 issue makes the following recs:
1. Screening for the above mutations to be done with all patients with aHUS prior to transplatation. I think that perhaps any non diarrheal related HUS should be screened as this might be the first hit.
2. Avoid Living related donation in such positive cases due to genetic transmission. Suggest a friend or spouse in such cases.
3. Studies have shown that aHUS MCP mutation can undergo kidney transplantation without increase risk of recurrence.
4. Anti CFH mutations might need pre emptive plasmapheresis, rituximab and steroids to lower the antibody levels.
5. The CFH and CFI mutations, the risk of recurrence is very high and transplant might be a risky procedure.
6. The options for CFH and CFI mutations might be combined liver-kidney transplantion along with TPE pre and post. Kidney alone with Pre and post TPE or kidney alone with eculizumab( anti complement agent).
All are only cases described, so no final decisions can be made. risk benefit has to be discussed with each case. Similar situations play part in C3 and CFB mutations as well. THBD mutations also are at risk but there is no data to do anything in these cases.
Does Nephrectomy of native kidneys help? Again, doesn't seem to be beneficial.
Check out these references:
http://www.ncbi.nlm.nih.gov/pubmed/20642678
http://www.ncbi.nlm.nih.gov/pubmed/20445192
http://www.ncbi.nlm.nih.gov/pubmed/20595690
Image source: http://www.profelis.org/amc/vorlesungen/immunologie/komplementsystem.html
Monday, August 2, 2010
TOPIC DISCUSSION: 25-OH Vitamin D and ESRD
 The KDOQI has now started recommending that we check inactivated Vitamin D levels in all CKD patients with hyperparathyrodism and if the level is <30, to start replacing them with either D2 50,000 units once a week for 3 months or D3 1000 units once a day for 3 months. This is the nutritional deficiency recommendation based on the general population. There have been many studies in the general population with effects on even none bone/muscle organs: cancer prevention, decreased renin, decreased cardiac disease and so forth.
The KDOQI has now started recommending that we check inactivated Vitamin D levels in all CKD patients with hyperparathyrodism and if the level is <30, to start replacing them with either D2 50,000 units once a week for 3 months or D3 1000 units once a day for 3 months. This is the nutritional deficiency recommendation based on the general population. There have been many studies in the general population with effects on even none bone/muscle organs: cancer prevention, decreased renin, decreased cardiac disease and so forth. 1. What are the implications of this in CKD and ESRD population?
2. Does it matter to replace inactivated vitamin D in ESRD patients?
3. Won't the inactivated D be more suppressed in our ESRD patients since we are giving them activated Vitamin D, sometimes in excess?
A recent study showed that lower levels of the inactivated Vitamin D was associated with increased mortality in HD patients but the administration of activated vitamin D to these patients decreased the mortality.
There is one study by Bert et al, that found that vitamin D3 was not as effective as activated vitamin D in decreasing pth levels in dialysis patients. Bone biopsies actually showed worsening of disease.
There is 1 alpha hydroxylation happening else where, mainly macrophages and other cells besides the kidney. Giving 25-OH might activated those cells to convert more and we can get extra renal activations.
But again, data is observational and there is no harm in giving nutritional supplemental vitamin D as long as the calcium and phosphorus are in good range.
The role of a combination of a calcimimetic and 25-0h vitamin D might be interesting to look at according to many experts. Well designed trials comparing both measures of just vitamin D alone vs vitamin D + calcimimetic vs activated vitamin D alone might be worth looking at.
Look at the below references
The first one is a nice review on all bone diseases in CKD( a nice table is in the article that summarizes a lot), good board prep table.
Kalantar-Zadeh K, Shah A, Duong U, Hechter RC, Dukkipati R, Kovesdy CP. Kidney Bone Disease and Mortality in CKD: Revisiting the role of Vitamin D, calcimimetics, alkaline phosphatase, and minerals. Kidney Int 2010:78 (suppl 117):S10-S21.
http://www.ncbi.nlm.nih.gov/pubmed/17687259
http://www.ncbi.nlm.nih.gov/pubmed/208439
IN THE NEWS:- MAYO CLINIC AND SOCIAL MEDIA
http://socialmedia.mayoclinic.org/
Check out the first center of Social Media by an University Medical Center. This is a true breakthrough.
I think that this will inspire many health care networks to have a more robust and prominent role of social media in health care.
Check out the first center of Social Media by an University Medical Center. This is a true breakthrough.
I think that this will inspire many health care networks to have a more robust and prominent role of social media in health care.
Subscribe to:
Posts (Atom)